 Masanobu NAKAYAMA
Masanobu NAKAYAMA
第一原理計算(解析編)
解析編では、第一原理計算で得られた一般的な出力をどのように加工するかを説明する。
(更新履歴)
何時までたってもできあがらないので、2005年2月28日にできたところまで公開
2006年1月19日。ひとまず完成。
2006年2月28日。文章を推敲
概要
(2005/1/5)
あなたが苦労して(というか、コンピューターの方が大分苦労をしたのだと思うのだけど)第一原理計算を終了させたとしても、実はまだまだ始まったばかり。これから大変なデータ解析をしないといけない。第一原理計算のデータ解析を経験すると分かると思うのだけど、結構、計算屋の仕事って単調で根気と忍耐力が必要な泥臭い作業なんだよね。
ところで、計算というやつは、現実を再現しているか否かに注目や議論(あるいはクレーム)が集中しがちで、結果はたくさん出て当然と思われる方もいらっしゃるが、そのままの生計算データを眺めてみても、実は得られる情報量ってそんなに多くないかもしれないと思う。だから、計算実験は、得られたデータを如何に有用なデータへ導くかということが重要なんでしょう、たぶん。
しかしここでは、そういった応用は述べず(まだまだ素人なので述べられない・・・)、ルーチンワーク的なデータの処理法を解説する予定。
トータルエネルギー1 単位
(2005/2/3)
基礎編でも述べたように、第一原理計算ではシュレディンガー方程式の固有値、すなわちトータルエネルギーが算出される。(一部、そんな値を出さないソフトもある)。このトータルエネルギーとは、ボルン・オッペンハイマー近似(基礎編)をしているから、原子核の情報を含まない、電子に由来するエネルギーである。だから、我々が普段見慣れている、エレメントのエンタルピーを基準(ゼロ)にした生成エンタルピーとは基準が違うので注意しなければならない。
また、おなじシュレディンガー方程式を解く第一原理計算でも、全電子法と擬ポテンシャル方では取り扱っている電子の数が全く違うので、同じ物質でも値は全く異なることに注意。くどいけど、LDA+Uの場合は、Uの値も比較するもの同士で一致させないとダメっす。
それから、プログラムによって異なる単位にも気をつけること。ここで、単位とは2つの意味があり、
1) いわゆるエネルギーの単位 (→換算)
2) 原子(イオン)個数の規格化
を考えなければならない。よくマニュアルを確認しておきましょう。
1)は、ちなみにWien2kの場合 Ry(リュードベリ)系、VASPの場合 eV系である。
2)は、時々間違う人が居るので注意。ちょっと詳しく述べてみよう。
固体に関するエネルギー表記は、慣習的に 〇〇kJ/molと書かれているけど、これでは1molの定義づけが非常にあいまいである。たとえばNaClのばあい普通Na 1個とCl 1個を合わせて1molと定義するので、正確には 〇〇kJ/mol per
NaClなのだろう。ところが、第一原理計算をするときに、Fm3m(岩塩型のF格子)で座標を入力したとなると、現実の計算はNa4Cl4の8原子で計算したことになる。つまり出力は〇〇kJ/mol per
Na4Cl4となるので注意。この場合は値を4で割ってあげなければならない。ところが、これは計算するソフトで違ってくる。Wien2kの場合は、入力ファイル作成プログラムがFm3mのシンメトリーを考慮してくれるので、実際の計算はPrimitive
Cellつまり単位格子1個NaClで計算してくれる(設定による)。一方、VASPの場合はそういった補助プログラムがないので8原子の結果を与える。ということなので、とっても気をつけてください。(あえて、kJ/molと書いたけど、こんな単位系で出力するプログラムは恐らく皆無?)
トータルエネルギー2 結合エネルギー
(2005/2/3)
ところで、トータルエネルギーがでてきたわけだが・・・冷静に考えると「だから何?」という状態ではなかろうか。これは僕の主観だけど、実際問題としてある物質のトータルエネルギー単体が出てきたところで材料科学的になにかの役に立つわけではないと思ったりする。現実には、データを加工して役に立つ数値にしなければならない。典型的な例として、ここでは結合と格子エネルギーの算出法を解説する。
結合エネルギーとか格子エネルギーとか
化学結合や格子のエネルギーは、学部の教科書にも載っている概念であるが、現実に紙と鉛筆で計算するのは難しい。しかし、計算によって求めることができれば(実験で求めるのは普通とても大変)それぞれの物質の結合エネルギーの比較なんかができて、材料物性の理解の助けになることは想像に難くない。第一原理計算ができる環境であれば、お手軽に計算することができる。しかし、単純にトータルエネルギーだけで比較できるのは組成が同じ化合物のペアに限られる(たとえば、面心格子のNaClと体心格子のNaClを比較するなどなど)。組成の異なる物質同士のトータルエネルギーを比較しようとしても、各原子の内殻電子のようにほとんど結合に寄与しない電子のエネルギーを含んでしまうので比較することはできない。ということで、組成が違う物質同士の結合エネルギーを比較するためにはデータに加工が必要。基本に振り返って考えて見る。
結合エネルギーは、たとえばAXという分子性化合物の場合、AとXという原子それぞれの単体のエネルギーをE(A), E(X)とし、AXという分子のエネルギーをE(AX)とした場合、
結合エネルギー = E(AX) - { E(A) + E(X) }
となる。注意しなければいけないのは符号で、ただのトータルエネルギーの場合は安定化するほど負の大きな値になるが、結合エネルギーの場合は、結合が強くなるほど正の大きな値になる。
図にすると下みたいな感じ・・・・。あまり図にする必要も無いかもしれないけれど・・・
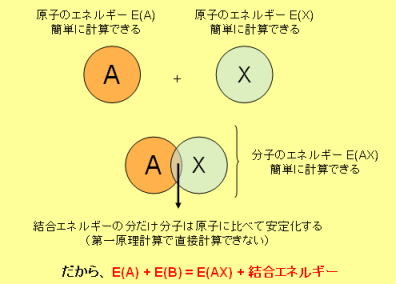
この要領が分かれば、色々なエネルギーを計算することができる。
イオンの結合エネルギーであれば
イオンの結合エネルギー(or 格子エネルギー) = E(AX)-E(A+)-E(X-)
この場合、電子を付け加えた・取り除いたイオンの計算なので、あらかじめ計算ソフトのマニュアルを参照しておくこと。ちなみこのエネルギーは結晶で考えた場合に、格子エネルギーに相当する。
更に、分子のイオン化エネルギーや電子親和力なら、
イオン化エネルギー = E(AX+) - E(AX)
電子親和力 = E(AX) - E(AX-)
最後に、結合や格子形成に原子やイオンの個数をよく考えて、各項に係数をつけることを忘れないように。
トータルエネルギー3 (擬似的な)生成エンタルピー
(2005/2/10)
化学反応における反応熱、あるいは生成エンタルピーもある程度求めることもできる。正確に言えば、求められる生成エンタルピーは熱力学の標準状態である1atm, 298Kではなく0atm,
0Kであることに注意すること(なので、擬生成エンタルピーと変にのたまっている)。でも、熱力学的な標準状態との差になるpV項は求められる値に比べてかなり小さいので(どの程度の精度を求めるかによるが)、大雑把な値を見積もるのには問題ない。ただし、反応に気体がからむと差が大きくなる傾向があるので注意。気体は自由度が大きいため圧力・温度による変化が大きく、値が大きく逸脱しがちである。
先に、どの程度の精度が出るのか実験値との対応表を見せる。物質名はちょっと記載できないのだが、申し訳ない。
メタルM, Nと酸素の二元系のデータをテーブル化したもの。実験値の出典は色々で、標準状態(298K,
1atm)での値だが、だいたい計算値のほうが数%ほど値が大きくなっている程度の系統的な誤差が生じる。これは基本的に先に述べた酸素分子の計算上の問題に由来していると考えられる。誤差を生じるものの、実験でこのデータテーブルを作るためにはすさまじい忍耐と時間が必要だが、第一原理計算でやれば、複雑な構造で無い限り1日もかからず大まかな熱力学データを得ることができる。

では、実際にはどうやって生成エンタルピーを計算するのだろうか?それは、実はとても簡単で生成エンタルピーの定義となる化学式を思い出せばよい(下の図)。熱力学では標準状態で単体である物質の相の生成エンタルピーを0に定義にしている。金属は水銀を除いて固体だし、酸素や水素なんかは気体分子になる。第一原理計算では、標準状態?が0K,
1atmだから酸素も固体になってしまいそうだけど、知りたいのは標準生成エンタルピーだから気体で考える。第一原理計算をやって、それぞれの単体のトータルエネルギーをだし、目標の化合物(下の図の場合はMNO、Nは窒素じゃないっす)のトータルエネルギーを算出すれば、
生成エンタルピー = -1*( E(MNO) - E(M) - E(N) -0.5*E(O2) )
なる式で算出できる。ただし、最初に-1をかけているのは、反応熱と生成エンタルピーの値は正負符号が逆のため。

☆正確には内部エネルギーを求めている。エンタルピーとの関係は H = U + pV。でも固体の場合はpV項は普通とても小さいし、熱力学データベースは普通定圧系・・・つまりギブス関数やエンタルピーとして与えられているので、ここでは(擬似的な)エンタルピーという歯切れの悪い表現をしている。・・・というか、熱力学の研究者にいい加減な人をほとんどみかけないので、こんなことをのたまっているだけで石をなげられそう・・・。
トータルエネルギー3 平衡電位の計算
(2005/2/20)
反応の(擬)エンタルピー(反応熱)も同様の方法で計算することができる。詳細は書くまでもないであろう。最後に、僕は電池屋なので、電池の平衡電位計算式を示す。さて、平衡電位の定義だが、これは化学ポテンシャルに由来する。ここで注意が必要なのがポテンシャルという言葉。エネルギーとは違う。前者(ポテンシャル)が示強変数なのに対して後者(エネルギー)は示量変数なのだ。・・・・って回りくどい言い方だねぇ。このことは、また後で例示しながら説明するとして・・・・もう一つ重要なのは、電位にはエントロピー項の影響を受けているということ。つまり系の乱雑さですね。第一原理計算ではエントロピー項を計算することが普通はできない(工夫をすれば振動と配置のエントロピーを求めることはできる。電子のエントロピーもできる。)のだが、実際には平衡電位を求める場合にはあまり問題にはならない(というのは、極端・・・・程度問題なんです)。それはエントロピー項が、常温(25℃)でかなり小さいからである。ということで、具体例を示しながら説明しよう。そして具体例は、例によってリチウムイオン電池なのだ。。。。
まずは、いわゆる平均電位を機械的に求めてみよう。これは近似であるので、色々問題は含まれる。
リチウムイオン電池の反応式として、商用にも使われているLiCoO2正極について、Li金属の酸化還元電位を基準にして述べる。この場合の反応式は、下記のように表される。
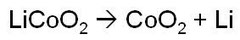
ところで、電位はギブス関数と
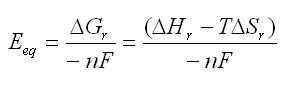
という関係がある。⊿G、⊿H、⊿Sはそれぞれギブス関数、エンタルピー、エントロピーに相当するのだが、先ほど述べたようにエントロピーはかなり小さいので無視できる。だから、エンタルピー項だけを用いて平衡電位が議論できる。またnは反応に関与する電子数であって、この場合はCo3+/4+ のレドックス系だから n = 1
となる。(ただし、固溶体生成反応なので厳密には取り扱いを変えないといけない。これは後で述べる。)あと、Fはファラデー定数ね。
さて、エンタルピーは前に言った方法で、擬似的に求めることができる。だから、LiCoO2、CoO2、Liの電子のトータルエネルギーは第一原理計算から、
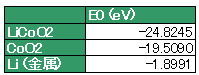
と与えられるから、反応エンタルピーは前のセクションから分かるように、
⊿H = E0(CoO2) + E0(Li) - E0(LiCoO2)
ここで、先ほど述べたようにn=1、T⊿S=0、単位はeV系なのでF=1なので、電位E=3.4165 Vとなる。実験値は3.8Vだから、過小評価していることが分かる。このズレは、交換相互作用項の近似の問題であることが指摘されている。ちなみに近似法はGGAを用いているが、あとでGGA+Uで修正した場合の例を示す予定。

最後に平均電位で求める電位の意味であるが、上の図のように計算をした2点間のトータルエネルギーを直線で結んだ時の組成に対する傾きを算出しているに過ぎない。これは、2相共存反応の場合の電位に相当する。固溶体反応が進行する場合は青い線で表されるように、実際のギブスエネルギーは直線で結んだ赤い線よりも小さくなる。
☆察しのいい人はお気づきだと思うが、なにしろ第一原理計算が苦手なのは、中間組成とか固溶体みたいなやつ。考えられる構造が複数あると困ってしまう。つまり、原理的にその思考法においてエントロピーがいつも0なんです(状態和を計算できない)。でも、たとえばヘンリーの法則とかラウールの法則とかを思い出してもらえば、ある程度は温度や固溶という概念をなんとか導入できる。更に、配列のエントロピーなら、工夫すればかなりの精度で計算可能。最近の僕の関心事はここらへん。振動のエントロピーも導入可能な概念らしいけど、そちらはマダマダ。。。時間がほしい。で、時間があったら、どこかで紹介したいとおもいつつ・・・・、今日はここでおしまい。
☆遷移金属酸化物のd電子は局在効果が大きいので、ハバードのU(オンサイトクーロン効果)を導入して補正することが最近のはやり?なのだが、実際その効果はどうなのであろうか? 大体の遷移金属の3dのUは4~5eVくらいになることが経験的に知られている(少なくとも中山の知る限り)。Uの値を様々な値に設定してLiCoO2系電極の平均電位を計算した結果を下の表に示す。ちなみに平均電位は約3.8Vくらいだから、さっきの経験値であるU=4~5eVくらいで値は一致しているように見える。実際、Uが3.5以上位になると中山の(乏しい)経験ではトータルエネルギーに限らずあまり結果に違いがない感触を得ている。なので、最近はUの値設定に必要以上にナーバスになる必要はないのではないかと・・・・。ただし、反応のように2つの状態を比較する時はUの値をそれぞれ等しくしないといけない。トータルエネルギーそのものが変化するため。もっとも、これには異論のある向きもあるだろう。実際、経験的な感想なのだし・・・

状態密度(DOS)
(2005/3/6)
DOSは、固体における電子の軌道状態図を表したものである。分子の軌道は量子力学の述べるところによりエネルギー値がセパレート(離散)しているわけだが、固体の場合は原子がアボガドロ数個、つまり軌道がアボガドロ数個スケールで存在するので、バンド(帯状)になる。(これ以上の詳しい説明は、いつか別のところでするかもしれないししないかもしれない。でもたった今、こ「そんなことは知っている」という人がバンド計算をするという前提で話すべきなんだと気づいたので、そういう話はおしまい。)
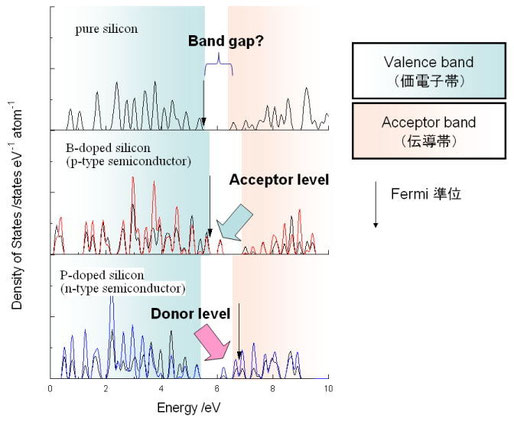
まずはDOSの表記の仕方についての注意からはじめよう。下の図は半導体シリコンのバンド計算の結果をまとめたものである。横軸は軌道レベル(エネルギー)、縦軸は状態密度である。
まず、横軸だがどこで0を取るかが問題となる。電子が詰まった一番高いエネルギーの値であるフェルミ準位を0にするか、それとも真空を基準にして0にするかの2つの方法が主流である。バンド計算のプログラムによって出力法が異なるので注意しないといけない(バンド計算のプログラムのオプションにもよる)。
ところでフェルミ準位の取り扱い方には注意が必要で、中山も実は完全に理解しているとは言いがたい。まず、実際的なバンド計算では計算の収束性を向上するために、電子の詰まり方をフェルミ分布やガウシアン関数によってsmearing(引き伸ばしている)ことがある。金属の場合はあまり問題がないのだが、バンドギャップが存在するケースでは、DOSの形状によって大きくフェルミ準位の位置を見誤ってしまうケースがあるので気をつけないといけない。また、バンドギャップをもつ化合物のフェルミ準位は「電子の染み出し」というあいまいな理由でバンドギャップの中央にあるとされるわけだが、普通のバンド計算のコードではそんな解釈はしないで、即物的に詰まっているところまでという値を返してくる。下の半導体シリコンのDOSがよい事例であろう。
最後に、真空基準のエネルギーの扱いには注意が必要。実験で電子を真空に持っていくエネルギーと考えてしまうと、現実には電子構造が違う表面を通過しないといけないから、バンド計算で出された値とは違ってくるらしい。これは僕もよく分からないので、分光スペクトルと軌道エネルギーをあわせて定量的に議論する時はよく調べてくだされ。
次に縦軸について、単位は基本的に軌道の数(密度)を表すわけなので、単位は例えばstatesという数になる。で、もし単位胞における軌道の数をカウントしている場合には、下のシリコンの場合Siが2個あるので、2Si
あたりということになる(図の例ではそういう単位のとり方はしていない)。また、先に述べたようにDOSというのはエネルギー軸に対して増えていく総状態数の微分という定義なので、eVで割っている単位になることに注意しよう。
それから2通りの縦軸のとり方があることを述べよう。ひとつは単位胞全てを対象にした出力で、もう一つは元素毎に出力する方法。前者は分かり易いだろうから、後者についてコメントする。バンド計算で定義される波動関数は固体全体に渡って(単位胞を越えて)広がる波だから、この波(軌道)はどこそこの元素に所属する波と定義することができない。ということで、元素毎に状態密度が求まると化学者としてはとても便利なのだが、本質的にそんなことはできない。なので次善を選択する。多くの場合、元素周りに適当な半径(これが、とてーもあいまい)で球(あるいはウィグナー・ザイツ胞)を切り出してくる。その中の軌道を投影して元素ごとのDOSを決定しているのだ。だから、適切な半径になっているかどうかは設定ファイルをみて気をつけないといけない。最近のコードは、適切な値を自動的(経験的に?)に決めるのもあるので、それに従ったほうが無難なことが多い。
最後に、DOSを求める際の注意点を一つ。計算プログラムにも夜が吐き出されたDOSファイルは格子緩和過程のDOSを、各格子緩和過程での平均としているケースがある(例えばvasp)。これは、おそらく第一原理分子動力学法用途のための措置だと思われる。したがって、最終的な結晶構造におけるDOSが知りたい場合は、格子緩和をしない計算をもう一回行って、最適結晶構造のDOSを計算しないといけない。
以上が、簡単な状態密度の説明。続いて具体例でDOSについてもう少しコメントを加えることにする。
具体例1) 半導体Siと、Siにボロン、リンを微量(でもないけど)ドープした場合の第一原理計算結果。
この図はトータルDOSではなくて、各原子ごとに投影したDOSを示している。シリコンは黒、ボロンは赤、リンは青です。それから縦軸は原子一つあたりに規格化していることに注意。本当はボロンやリンは極少量なんですが、規格化のために軌道の数がシリコン並みにたくさんあるように見えてしまってる。で、シリコンの場合は大きな?バンドギャップが存在しているわけだけど、ボロンを入れるとボロンのアクセプターレベルの、リンを入れるとドナーレベルの軌道がそれぞれ現れる。Pの場合にフェルミ準位が大幅にずれてしまったのは、よく分からないけどさっき述べたsmearingのせいだと思われる。格子内のリンの数が多い(本当は超微量のはずなのに、この計算は本ホームページ用に10分で行った計算なので7個のシリコンに対して1個のリンが入っている)ためにずれているのかもしれない。
さて、お気づきかもしれないがバンドギャップのところに ?マークがある。実は、バンドギャップの見積もりは一電子近似の密度汎関数法では非常にナーバスな問題であって、あまりうまく現実を再現できないことが知られている。いわゆるLDAによる近似では、バンドギャップは実験値と比べると常に過小評価され、実験値と一致しない。シリコンのバンドギャップの実験値は、1.17
eV、これに対してLDAをやるとバンドギャップは、0.4~0.5 eV程度の値となる。LDAではいつも過小評価になるけど、なんというか計算する系によってその程度は異なるからややこしい。
正しいかどうか知らないけど、下の計算例はGGAでやっている。そうすると割りといいんじゃない?という値になっている(けど半導体のことはよく知らない)。結局、電子交換相互作用が問題になっていて、その程度が大きい酸化物の場合はGGAで計算してもバンドギャップは再現しない。基本的に1電子近似であるDFTではダメだという意見もあれば、最近はLDA+UとかSIC、GWなどで結構何とかなっていたりする。
また、バンドギャップの見積もり方として電子の授受の観点から、トータルエネルギーで見積もる方法もあり、こちらのほうがsmearingの影響など受けないので正確である。系を中性にした時のトータルエネルギーをE(N)とする。ここでNは格子中の電子の総数を意味する。格子に1個電子を入れた系と1個電子を抜いた系のトータルエネルギーをそれぞれE(N+1),
E(N-1)とした時、バンドギャップEgapは、Egap = E(N+1) + E(N-1) -2E(N) で表される。
☆バンド構造をk空間上で表したウネウネした曲線がのた打ち回るバンド図(個人的にはスパゲッティーダイアグラムと呼んでいる)で電子構造を解析できるツワモノも居るが、僕はそこまではちょっと・・・・というレベルなので、その微分曲線であるDOSの方がなじめる。
具体例2) リチウムイオン電池電極材料のLiCoO2のDOS。黒線がCo,
青がO、赤がLiを表す。一見して分かるように、コバルトと酸素のDOSの形が酷似していることが分かる。LCAO流に解釈すれば、これは両者の軌道が重なって混成軌道を形成していることを意味している。端的に言えば、CoとOが強い共有結合を形成していることを意味している。また、-2eV 以下では酸素の軌道の寄与が大きくなっており、O 2pバンドと解釈される。一方、-2~0
eVに形成されているのは酸素と6配位したCoのt2g軌道、3~4eVにあるのはeg軌道に相当する。t2g軌道はフェルミレベル以下に形成されているから、6個のd電子がCoの軌道を占有していることになり、Coは3価であることが分かる。またバンドがアップ・ダウンスピンで対称形なので、low spin錯体になっていることも分かる。

電子密度解析
(2005/6/6) 加筆(2005/1/24)
波動関数を2倍すると電子密度になる。だからシュレディンガー方程式を解くと簡単にデータを得ることができる。多くの第一原理計算では標準出力として電子密度の結果をファイルに吐き出してくれる。分子の電子密度を、視覚化するととても美しいが、現実的には有用な情報として抽出して議論するのは目的にもよるが難しい。そこで、いくつか中山の知っているやり方(あまり引き出しはない)を紹介しよう。なお、ビジュアル化するプログラムとしては、フリーではVENUSがある。直感的に操作でき、対応フォーマットが多いので使い易いと思う。以下の図は、すべてVENUSで出力されている。
1.結果をそのまま見る。(共有結合性やイオン結合性の議論)

上はオリビン型構造を有する燐酸コバルトリチウム(LiCoPO4)の電子密度をWien2kで計算した結果である。電子密度図を見て分かるように、PO4ユニット(図中の赤い丸で囲まれた部分)は、PとOの電子密度が大きく重なっている(共有結合)。一方、青い丸で囲まれたCoO6はほとんどCoとOの間で電子雲の重なりがないので、イオン結合性が強いと言えよう。このような極端な例では、どの結合が共有結合性で、イオン結合性なのかが分かる。しかし、あくまでも視覚上の結果であるので電子雲がくっついている、いないの議論でイオン性云々を議論するのは危険である。
2.原子軌道由来の電子密度から差をとる。
1)水素 H2
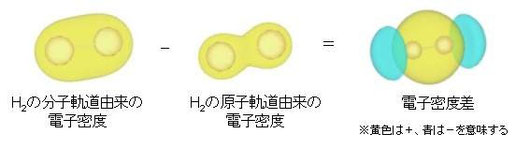
上はH2の分子軌道由来の電子密度から原子軌道由来のそれで引いた時の差密度を示している。計算はvaspで行っている。共有結合部分で電子が増えていることがわかるであろう。また反結合σ*の電子が減っていることが分かる
(これはLCAO法で考えると分かり易い。計算は平面波基底だけど。)
2)コバルト酸リチウム LiCoO2

図示すると全体量としては分かりにくいが、(1)コバルトの電子密度が減少、(2)一方で酸素は電子密度が増加している。したがって、コバルトはカチオン化、酸素はアニオン化していることが分かる。イオン性結晶の場合は、水素の例のように共有結合云々の程度問題を議論するのは難しい。(電子の動きは中性原子→イオンになる過程で大きいため)
3.反応前後の電子密度差を調べる。
リチウムイオン電池の電極反応のように反応がトポタクティックに進行する場合、反応前と反応後の電子密度を引き算することで、酸化還元の電子移動解析をすることが出来る。通常は、トポタクティックといえども格子の変化があるが、これは完全に同一であるという仮定をしないといけない。
LiCoO2 - CoO2
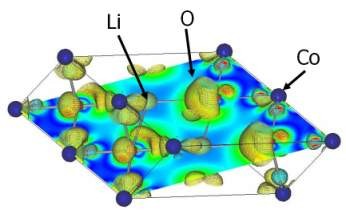
形式的な電池反応(放電)は、 Co4+O2-2 + Li+ ->
Li+Co3+O2-2 だから、予想図式的にはCo廻りで電子が増減するはずだが、実際には電子の動きがかなり複雑なことがわかる。Coは軌道によって増減の度合いが変化しており、egではむしろ減少していたりする。またOの電子も変化しており、静電場の影響を受け易いリチウム廻りで大きく電子が増加していることが分かる。これは、ローカルにイオン性の強いリチウム正電荷を中性化しようとする遮蔽効果が作用しているためだと考えられる。
4.ELF(Electron Localization Function)解析をする
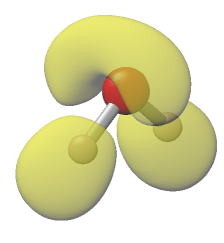
ELFは0から1で定義される量で、ELF解析をすることで、対電子または不対電子の分布を知ることができる。つまり、共有結合や孤立電子対になっている場所、ダングリングボンドの存在をビジュアル化することができる。上の図は水分子の例。酸素上に広がった孤立電子対(lone
pair)の分布がよく分かる。H原子上の分布も結合に沿った方向で広がっているように見える。ということで、化学結合や反応サイトを検討するときには有効な手法である。ただし、ELFはその定義に電子密度のラプラシアンの2乗を含むので、ちょっとしたノイズ(?)も大げさに反映してしまう。なので、電子密度を求める時はかなり精度の高い計算をしないといけない。
詳しくは下記ホームページか文献を参照のこと。この文章を書いている時点で中山もよく分かっていないので、間違っているかも知れません。
・ http://www.cpfs.mpg.de/ELF/
・ Becke, A. D.; Edgecombe, K. E. J. Chem. Phys. 1990, 92, 539.
5.スピン分布を調べる。
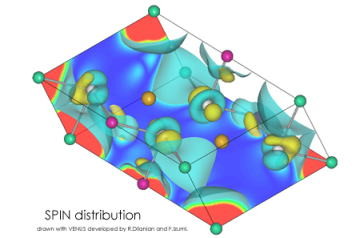
スピン分極をさせて第一原理計算をさせると、アップスピン(up spin)とダウンスピン(down
spin)の電子密度をそれぞれ別に計算してくれる。これまではアップとダウンスピンの和(つまりトータル電子密度)を可視化していたわけだが、アップとダウンの差を可視化することもできる。それを行なったのが上の図。黄色い等電子面と青い等電子面はそれぞれ正負の別を示している。磁性の研究をする人なんかは重宝する結果なのかもしれないが、中山はこの分野に詳しくないのでイマイチ、ピンとこない。しかし、ハイスピン錯体の時は、後述の球面積分法を使うと、この結果からイオン価数を見積もることができるので便利。
6.球面積分(チャージインテグレーション)をする
上で述べてきたように電子密度をビジュアル化すると化学結合などを直感的に理解できるので、非常に便利なんだけど、やはり定量性には欠けてしまう。 定量性を含めた議論には、原子核(イオン芯)から半径方向に電子密度を積分(球面積分)してプロットするとよい。 実は電子密度以外にも、差密度(例えば原子軌道関数との差密度)やup-downスピンの差密度など、なんでもいいから球面インテグレーションすると分かり易い情報が得られると思う。
例として(1)遷移金属酸化物中の酸化物イオンの電子密度を球面積分したものを下に示してある。大体、酸化物イオンのイオン半径(1.2~1.4Å)付近で電子数が7~8になっていて、電子密度の変化がすこしなだらかになっていることが分かるであろう。この電子密度は擬ポテンシャル法で求めているので、価電子だけをカウントしているから、中性の酸素だと6電子持っているはずである。従って、電子数が中性の時よりも多いので、アニオン化していることが分かるだろう。この方法を差密度に適用すると、電子が動いた量が一目瞭然なので重宝する。特に遷移金属のd電子などは軌道によって電子が増えたり減ったりしているので、トータルでどれだけの電子が動いているかよく分からなくなることがあるのでなおさらである。

次の例(下図)は(2)クロムを含む複合酸化物中のクロム周りのスピン・インテグレーションである。スピン・インテグレーションとは、アップスピンとダウンスピンの差を球面積分した結果である。電子密度図だけではイオン価数を調べることが難しいが、球面積分すると、約1~2Åの範囲でプラトーが現れる。このときの値はハイスピン錯体の場合(クロムの場合はそんなことを考えなくても、スピンはみんなアップになるんだけど)、d電子の値を考慮するとCr3+では3に、Cr4+の場合は2になるはずである。 Cr4+は中々とりえない価数なんだけど、図の赤線で示されるようにある種の複合酸化物ではCr4+になっていることが分かる。

構造緩和(イオン緩和)の結果を利用する
構造緩和した結果は、実験(たとえば回折法)で得られた構造と照らし合わせて、計算の妥当性をしらべるのが一番ポピュラーに使われる。この場合格子定数や分率座標を細かくチェックするのが解析の第一歩。 ただし複雑な構造だったり、特にトポロジーの変化なんかを知りたい場合は、得られた結果から改めて回折パターンにしてしまうのが手っ取り早く比較できる方法である。 回折パターンはあるけど構造が分からない時なんかは、候補を絞ることもできる。
元素の部分置換や欠陥などを発生させた第一原理計算の結果は、平均構造だけがわかる回折法と違って局所構造を与えてくれる(第一原理計算は逆に平均構造をだすのが難しいのだけど・・・)。ここでいう局所構造とは、結晶学的に等価なサイトにある原子でも、元素種や欠陥種によって構造が歪んでいたりする情報を得ることができることを指している。こういう結果の解析には、地道に結合長や結合角の分布を調べていくので結構大変なんだが、(おそらく)重要。
(応用例) Bond Valence Sum (BVS) によるイオン伝導経路の可視化。
この方法は、別に第一原理計算の結果に限っての方法と言うわけではないのだが、イオンの配列が分かれば、任意の空間上の一点にイオンを置いたときのBVS値というのを計算することができる。(BVSが分からない人は、下記論文を参照してください)。酸化物中のリチウムイオン伝導を考えた場合、リチウムは1価のカチオンとして存在するので、BVSが1に近い値をとる空間をリチウムは伝導すると考えられる。それを行なったのが下の図で、ペロブスカイト中でのリチウムイオン伝導経路を示している。

*BVSの参考文献 --> I. D. Brown, Acta Crystallogr. Sect. B 48, 553 (1992) とか。
あとがき
途中、大分さぼってしまったがとりあえず、中山が(今のところ)知っている第一原理計算の結果をシンプルに解析する方法はここら辺でおわり。(注:2006/1/24現在 まだ未筆部分あり)。 もちろん第一原理計算の結果を更に上手に使って、色々な物性値を出す方法は他にも沢山あるけど、それを説明するのは骨が折れることなので、論文を参照して欲しい。
たとえば、
・ 電子遷移状態などを計算する。 --> 内殻に電子空孔を発生させる
・ 原子・イオンの格子内拡散現象を取り扱う。
--> たとえば nudged elastic band method なんかがある。
・ フォノン・ディスパージョンを調べる
・ 合金や固溶系の相平衡を調べる
--> 第一原理計算+モンテカルロ法など。
・ 有限温度での分子運動など
--> 第一原理計算分子動力学法。
工夫次第で実験と同じくいろいろなことを調べられる(と思う)ので是非i色々と試してもらいたい。